
Researchers Use New Sequencing Methods To Study Protein Binding and Evolution
By Hannah Hirou
March 11, 2020 at 8:00 am
UC Santa Barbara researchers Justin Yoo and Michelle O’Malley, along with prior UCSB chemical engineering professor Patrick S. Daugherty, have innovatively reimagined the way that protein sequencing is done. In a recent study, they investigated the effects that different protein mutations in an adenosine receptor, a type of G-protein-coupled receptor (GPCR), have on ligand binding in humans.
GPCRs consist of a massive family of cell-surface receptors. When a ligand, such as a drug, binds to a certain corresponding GPCR, it triggers cascades of interacting secondary-messenger proteins to activate other biological pathways. These receptors play a key role in understanding how the body functions and reacts. GPCRs are involved in pathways such as heart function, vision and drug interactions.
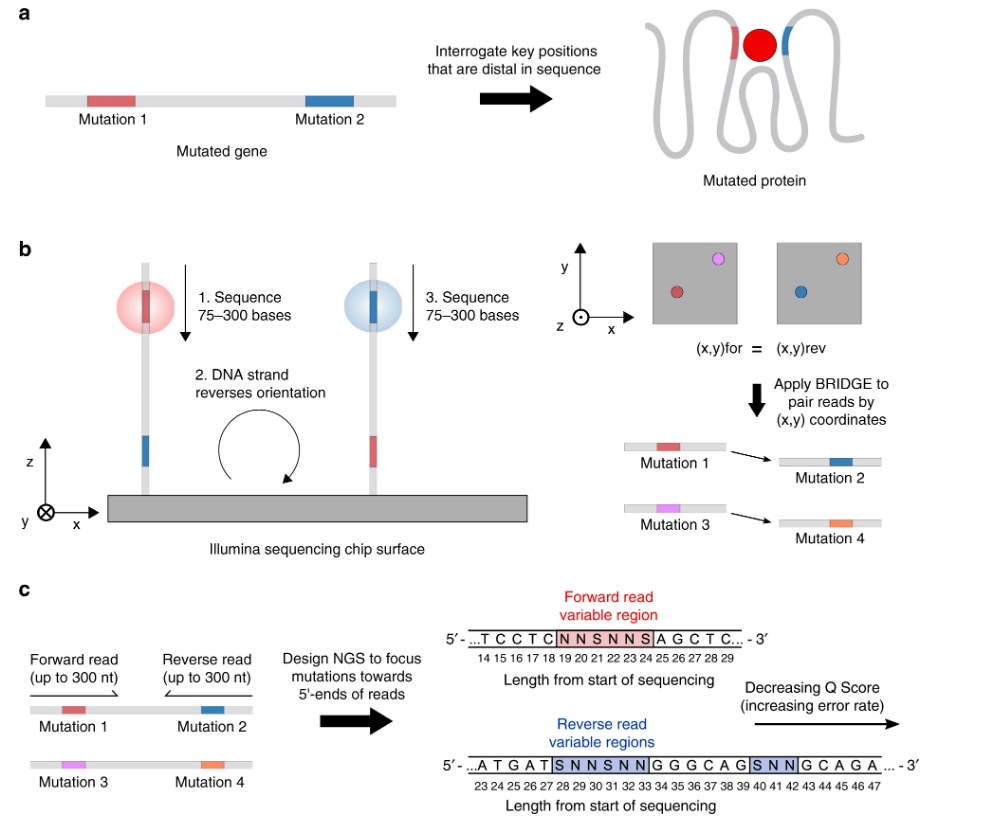
The paper, published in Nature Communications, addresses something known as B.R.I.D.G.E., or bridge reads in distal sites within genes, which was one of the methods used in this study. B.R.I.D.G.E. is able to read nonoverlapping pairs of DNA and makes the study of mutations more accessible. Traditional methods of DNA sequencing require overlapping strands of DNA before the sequence can be read, which limits the identification of amino acid substitutions (i.e. mutations), resulting in protein mutations, that occur close to each other.
“Part of the innovation here with this B.R.I.D.G.E. technology is that we are stitching together parts in a protein that would normally not be analyzed in the way that we did,” said O’Malley, an associate professor in the Department of Chemical Engineering.
B.R.I.D.G.E. takes advantage of Illumina technology, which finds individual base pairs in a DNA sequence and creates numerous short stretches of contiguous DNA base pairs that can patch together regions of proteins. This allows information on protein changes to be gathered comprehensively rather than in localized parts.
“Deep mutational scanning is this method that allows for all of the protein variants and mutations to contribute to the ultimate protein that is selected at the end of a screening,” O’Malley explained.
This accessibility is key in exploring epistasis, which is a key question in protein engineering that pertains to evolutionary relationships between gene mutations and protein binding.
“If you have a protein that you want to evolve for a certain function, there are lots of different ways to get there. Traditionally, a lot of people have made mutations to the protein to mimic how evolution and natural selection would work,” O’Malley stated.
She used the metaphor, “finding a needle in a haystack,” to explain how protein sequencing is used to find the needle if the needle were the mutation.
“Traditionally, [with] most techniques, if you find the needle in the haystack, you are happy. But you don’t know what other needles you might have left in the haystack or what else was in the haystack,” she said.
Examining a protein haystack requires multiple tools. Yoo, a former UCSB graduate student now based in Seattle, was the architect of the brilliant strategy that combined multiple sequencing processes.
“We combined next-generation sequences approaches to state-of-the-art Illumina technologies … with flow cytometry and cell-based screening that, in a more traditional sense, made it possible to actually screen through,” O’Malley elaborated.
This high throughput sequencing technology is useful in studying protein evolution. Once the researchers had their innovative protein sequencing method, they decided to test it. To do this, they engineered mutated versions of the well-studied human adenosine receptor, or the A2aR, to examine the potential effects on binding affinity to the receptor.
Five sites on the receptor were mutated based on other studies that suggested this had an effect on the binding affinity of the receptor to its ligand. To study this, Yoo “set up this library to basically create millions of variants that took these five places in the proteins and subbed out all the possible amino acids,” O’Malley added.
Coupled with sequencing, Yoo’s library allowed the team to know exactly which variants were winning.
Among the noteworthy things that Yoo saw were “mutants that, by themselves, were actually worse for the protein function, [but] when they co-occurred, were actually sometimes beneficial,” O’Malley said.
It seems counterintuitive that, for example, one protein mutation is bad but two are good. Given this, it is not simple to predict how mutations will affect protein function. It is comparable to how a negative number multiplied by a positive number results in a negative, yet two negative numbers multiplied by each other results in a positive product.
Epistasis relates to the interactions of genes with one another, and in this context, mutations have the potential to positively affect ligand-binding affinity.
“The additive effect of these five mutations were actually really hard to predict. That is this term epistasis that, in the paper, is the tracking of the co-occurrence and net benefit or net drawback of these co-occurred mutations,” O’Malley described.
Mutations were made at varying distances in the DNA sequences to see if this would have any effect on the ligand binding. Interestingly, the results of the study seemed to indicate that “mutations that were further away had an additive effect that was greatest,” O’Malley summarized.
While B.R.I.D.G.E. is a clever method that makes protein sequencing potentially more accessible to studying mutations, it does have its drawbacks. Namely, it cannot be used for sequences longer than 600 base pairs. However, the paper ends on a hopeful note that future advancements in technology can help improve this method and thus reveal more information on protein evolution.
Normal chromosome translocation inversion addition and deletion…changes in chromosome structure..hi to that transfer your at UCSB..you know Nelson Computers is out talking about Moore’s law.